
 Fact-checked by
Fact-checked by  Discussion
Discussion
Hemophilia overview
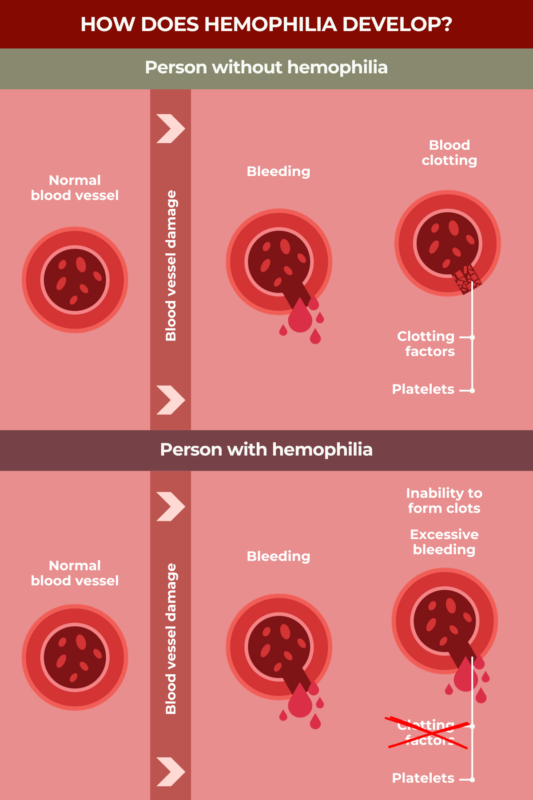
Hemophilia is a rare disorder in which a person’s blood is not able to clot properly, resulting in unusually easy and prolonged bleeding.
Within the blood, proteins called clotting factors normally work together to help form blood clots to prevent excessive bleeding. In people with hemophilia, one of these proteins either does not work properly or not enough of it is made, resulting in a clotting factor deficiency.
There are several types of hemophilia, each characterized by a deficiency, or lack, of a specific clotting factor protein. The most common form is hemophilia A, followed by hemophilia B. Both of these types primarily impact boys and men. Almost all cases of hemophilia are caused by mutations in a gene that provides instructions for making a clotting factor protein.
No cure exists for hemophilia, but there are a number of treatments available that can help to minimize and control bleeding in patients. While historically most people with hemophilia have not survived to adulthood, with modern medical care, individuals born with the condition in the 21st century now have a near-normal life expectancy.
Hemophilia causes
In hemophilia, one of the clotting factor proteins normally needed for the blood to clot does not work properly, either because the protein itself is abnormal, or because too little or none of it is being produced.
In the vast majority of cases, hemophilia is caused by mutations in a gene that provides instructions to make a specific clotting factor protein. These mutations are present from the moment a fetus is conceived.
The specific gene that is mutated determines the type of hemophilia a person has:
- Hemophilia A is caused by mutations in the F8 gene that provides instructions to make clotting factor VIII, known as FVIII.
- Hemophilia B is caused by mutations in the F9 gene that encodes clotting factor IX, called FIX.
- Hemophilia C is caused by mutations in the F11 gene that encodes factor XI, known as FXI.
In about two-thirds of cases, hemophilia-causing mutations are inherited from a person’s biological parents; in the other third or so of cases, patients are born with hemophilia despite having no family history of the disease. These instances generally are due to spontaneous mutations that arise during the early development of an individual sperm or egg cell that was used to conceive the child.
Because the F8 and F9 genes are both located on the sex-determining X chromosome, hemophilia types A and B predominantly affect boys and men. That is because male children only inherit one X chromosome from their mother. Female patients who inherit two X chromosomes, one from their mother and one from their father, may develop the condition if they inherit two X chromosomes containing faulty F8 or F9 genes, or if they receive a faulty copy and the other gene copy is present in the X chromosome that becomes inactivated. X-inactivation is a natural process in which one of the X chromosomes is shut down; this process has evolved in women to compensate for the extra X chromosome. In hemophilia C, the F11 gene is not on a sex-determining chromosome. Thus, type C affects people of all sexes at roughly equal rates.
People who carry one copy of a gene with a hemophilia-causing mutation, and a second healthy copy of the gene, usually won’t develop overt symptoms of the disease. However, because they may still pass the disease-causing mutation on to their biological children, these individuals are referred to as carriers. In hemophilia A and B, carriers are exclusively female individuals, while in hemophilia C, both female and male individuals can be disease carriers.
Types of hemophilia
There are three main types of hemophilia:
- Hemophilia type A, sometimes called classic hemophilia, is the most common. It’s caused by a deficiency in FVIII.
- Hemophilia type B, also known as Christmas Disease, is the second-most common type. It is caused by a deficiency in FIX.
- Hemophilia type C, also called Rosenthal disease, is notably rarer than the other two types, and is caused by a deficiency in FXI.
There are several categories of hemophilia severity based on the activity levels of the missing clotting factor. Clotting factor activity usually is expressed as a percentage, where 100% theoretically denotes the activity seen in a healthy person, though activity anywhere from 50% to 150% is generally considered normal.
- In mild hemophilia, clotting factor activity is higher than 5%, but lower than 40%.
- In moderate hemophilia, clotting factor activity ranges from 1% to 5%.
- In severe hemophilia, clotting factor activity is lower than 1%.
People with severe hemophilia commonly have notable bleeding problems during infancy, so they are usually diagnosed early in life. In severe hemophilia, it’s common for patients to experience joint bleeds, as well as spontaneous bleeding episodes — bleeds that occur in the absence of a clear cause. By contrast, patients with mild or moderate hemophilia often aren’t diagnosed until later in childhood or even adulthood, as they usually won’t experience abnormal bleeding except after an injury or trauma.
Although there can be a lot of variability from person to person, in general, studies suggest that hemophilia A is usually more severe than hemophilia B, while hemophilia C is typically much less severe than the other two forms.
Acquired hemophilia
In very rare cases, hemophilia can be acquired, meaning that it develops during a person’s lifetime, rather than due to a genetic mutation that’s present at birth. In these instances, the disease arises due to a malfunction of the immune system that causes it to wrongly target and destroy the body’s own healthy clotting factor proteins, most often FVIII.
In terms of disease severity, it can vary widely in people with acquired hemophilia. While some patients experience frequent and serious bleeds, some even life-threatening, others have relatively mild symptoms and do not need any specific treatment.
Hemophilia symptoms
The main symptom of hemophilia is bleeding that occurs abnormally easily and lasts an unusually long period of time. People with hemophilia may experience external bleeding, where blood loss occurs outside the body, or internal bleeding, which takes place within the body.
Common forms of external bleeding in hemophilia include:
- frequent nosebleeds that develop without a clear cause
- unexplained bleeding in the mouth, or excessive blood loss, after a dental procedure, such as tooth extraction
- heavy bleeding from a small injury, or heavy recurrent bleeding from a wound that had previously stopped bleeding
- unusual bleeding following a medical procedure that punctures the skin, such as vaccination or circumcision.
Manifestations of internal bleeding in hemophilia typically vary by patient, but may include:
- frequent or unexplained bruising
- bleeding into joints, which can cause joints to swell and be less mobile
- bleeding into muscles, which can cause significant blood loss and other complications
- pain, swelling, tenderness and/or rigidity in the abdomen
- blood in the stool or urine
- vomiting or coughing up blood
- bleeding in the brain, which can cause symptoms like double vision, weakness, headache, and seizures.
The most serious type of bleeding in hemophilia is generally considered to be bleeding in the brain.
Potential complications
Recurrent bleeding in the joints can lead to a condition called hemophilic arthropathy, which is marked by permanent and irreversible joint damage, joint pain, and limited mobility.
Bleeds in large muscles can cause serious blood loss, while bleeding into confined muscles like those in the forearms and calves can cause a painful condition called compartment syndrome due to increased pressure in and around muscles. Muscle bleeds also can put pressure on nerves, leading to symptoms like numbness and tingling.
People with hemophilia are at higher risk of having low bone mineral density, which can set the stage for conditions like osteoporosis to develop. In osteoporosis, bones become weaker and more prone to break, or fracture.
Hemophilia diagnosis
The main way of definitively diagnosing hemophilia is via clotting factor tests, also called factor assays. These are blood tests that measure the activity of individual clotting proteins. By identifying which clotting factor is deficient and to what level, these tests can determine the type, as well as the severity, of hemophilia.
Other screening tests, which can be used to assess overall blood health and clotting ability, may be useful for ruling out other conditions that can cause excessive bleeding. These include a complete blood count, known as a CBC, the activated partial thromboplastin time or APTT test, the prothrombin time or PTT test, and the fibrinogen test.
Because most cases of hemophilia are caused by mutations inherited from a person’s biological parents, establishing whether an individual has a family history of hemophilia can be crucial in making the diagnosis. For expectant parents at risk of having a child with hemophilia, genetic testing can be performed during pregnancy to determine whether the baby will have the disease.
Hemophilia treatment
The main treatments used in hemophilia A and B are factor replacement therapies that contain a working version of the missing clotting factor. Such treatments can be given to patients on demand, to control active bleeds, or prophylactically, meaning preventively, to reduce the frequency and the risk of bleeds. Factor replacement therapies can be plasma-derived, meaning they contain clotting factors isolated from the plasma of people without the disease; or recombinant, meaning they contain lab-made versions of those proteins.
Approved factor replacement therapies for hemophilia A include:
- Advate (octocog alfa)
- Adynovate (rurioctocog alfa pegol)
- Afstyla (lonoctocog alfa)
- Alphanate (human antihemophilic factor/von Willebrand factor complex)
- Altuviiio (efanesoctocog alfa)
- Eloctate (efmoroctocog alfa)
- Esperoct (turoctocog alfa pegol)
- Humate-P (human antihemophilic factor/von Willebrand factor complex)
- Jivi (damoctocog alfa pegol)
- Koāte (human antihemophilic factor)
- Kovaltry (octocog alfa)
- Novoeight (turoctocog alfa)
- Nuwiq (simoctocog alfa)
- Wilate (human von Willebrand factor/coagulation factor VIII complex)
- Xyntha (moroctocog alfa)
Approved factor replacement therapies for hemophilia B include:
- AlphaNine SD (human coagulation factor IX)
- Alprolix (eftrenonacog alfa)
- BeneFIX (nonacog alfa)
- Idelvion (albutrepenonacog alfa)
- Ixinity (trenonacog alfa)
- Profilnine (factor IX complex)
- Rebinyn (nonacog beta pegol)
- Rixubis (nonacog gamma)
Standard half-life recombinant factor replacement therapies contain a version of a clotting factor that’s similar or identical to the naturally-occurring version of the protein. Newer extended half-life or EHL recombinant replacement therapies contain a version of the protein that’s been modified so that the medication lasts longer in the body, allowing for less frequent dosing. Needing fewer doses can help to ease the treatment burden for some patients.
Apart from factor replacement therapies, other treatments that promote blood clotting through mechanisms that do not involve replacing the missing clotting factors can be used to prevent bleeds in hemophilia. Among these nonfactor therapies are the antibody-based therapies Hemlibra (emicizumab-kxwh), Hympavzi (marstacimab-hncq), and Alhemo (concizumab-mtci), as well as the RNA-based therapy Qfitlia (fitusiran).
Another class of treatments, called bypassing agents, also are available to treat certain hemophilia patients who have developed inhibitors, or neutralizing antibodies, against clotting factors supplied by factor replacement therapies. Bypassing agents approved for the treatment of hemophilia A and B include: FEIBA (factor eight inhibitor bypassing activity), NovoSeven RT (eptacog alfa [activated]), and Sevenfact (eptacog beta).
Gene therapies also have become available in recent years for some individuals with hemophilia. These one-time treatments are designed to deliver a working version of the gene whose mutation causes hemophilia, thus restoring the body’s ability to produce a functional clotting factor protein. Roctavian (valoctocogene roxaparvovec-rvox) is a gene therapy now available for hemophilia A, while Hemgenix (etranacogene dezaparvovec-drlb) can be used for hemophilia B.
There are also adjunctive or supportive treatments that may be used as an add-on to other therapies or in specific circumstances to help manage bleeding in people with hemophilia. Such therapies include antifibrinolytics and DDAVP (desmopressin acetate).
How common is hemophilia?
Worldwide, it’s estimated that about about 836,000 people may be living with some form of hemophilia, with as many as 33,000 male patients living in the U.S.
Hemophilia A, the most common type, is estimated to occur in about one of every 5,000 live male births. The next most common, hemophilia B, occurs in approximately one of every 25,000 live male births.
Both of these types of hemophilia affect people of all racial and ethnic backgrounds, and at approximately equal rates. These disorders do tend to be more common in cultures where consanguineous marriage, or marriage between two people who are genetically related, is more common.
Hemophilia C affects about one of every 100,000 individuals. This type of hemophilia affects both males and females at equal rates, and it is more common among Ashkenazi Jews, affecting about 8% of people in this ethnic group.
Acquired hemophilia, which is caused by a malfunction of the immune system rather by genetic mutations, is very rare — affecting an estimated 1.5 of every million people.
This rare form of hemophilia mostly affects people older than 65. About half of patients have other underlying health conditions, like other immune disorders, cancer, or certain skin or lung diseases.
Hemophilia inhibitors
Factor replacement therapies, in which a working version of the missing clotting factor is administered to patients to prevent or control bleeds, is considered the gold standard treatment for hemophilia.
But while replacement therapies can be effective for controlling bleeding, in some instances, the body’s immune system can mistake the delivered clotting factor for an infectious threat. When this happens, immune cells make antibodies that bind to the clotting factor, neutralizing it. These antibodies are referred to as inhibitors, because they prevent replacement therapies from working effectively.
If a person with hemophilia does not respond as expected to replacement therapies, or if a replacement therapy unexpectedly stops working as well as it had been, it may be a sign that the patient has developed inhibitors. It’s broadly recommended that patients be routinely tested for the presence of inhibitors in these situations.
Inhibitors are most common in hemophilia A, with estimates indicating that up to 1 in 5 people with hemophilia A will develop inhibitors at some point. Among people with hemophilia B, this is a less common problem: About three of every 100 patients with this type will develop inhibitors.
Other factors associated with an increased risk of developing inhibitors include:
- higher or more frequent doses of replacement therapies
- greater lifetime use of replacement therapies
- Black race or Hispanic ethnicity
- a family history of inhibitors.
In hemophilia patients who develop inhibitors, bleeding can be managed with bypassing agents. These are medications that can promote blood clotting without requiring the presence of the clotting factors that are missing or being targeted by an inhibitor.
Immune tolerance induction, or ITI, a treatment strategy that aims to re-educate a patient’s immune system so it no longer sees the delivered clotting factors as a threat and stops making inhibitors against them, also may be used in patients with inhibitors. With ITI, patients are generally exposed to a large amount of clotting factors over the span of months or even years.
Hemophilia life expectancy
For most of human history, hemophilia was regarded as a fatal disease. In the 1960s, the majority of people born with severe hemophilia did not live to adulthood.
Modern treatments have dramatically changed these outcomes, however. Nowadays, people with hemophilia who are diagnosed and started on treatment as children generally have a life expectancy that is comparable to that of the general population, according to estimates from the World Federation of Hemophilia (WFH).
A patient’s prognosis may vary somewhat depending on the severity of disease, but today even children with severe hemophilia are generally expected to have a normal life expectancy, if they are started on treatment early.
Based on the same WFH estimates, the life expectancy for men already living with hemophilia is about 10 years shorter than that for men without hemophilia. These estimates, however, are at least partly driven by other complications, particularly high rates of blood-borne infections transmitted to hemophilia patients via certain medications in the 1980s.
With modern screening techniques, the risk of getting an infection from hemophilia treatments nowadays is slim to none, which has helped to improve outcomes for patients diagnosed and treated in the 21st century.
Hemophilia C is generally much milder than hemophilia A or B. Typically people with hemophilia C don’t have a decreased life expectancy.
In rare acquired forms of hemophilia, the risk of mortality has been suggested to be highest in individuals who are elderly or have other health problems like cancer.
Hemophilia News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
Related articles
-
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
 Discussion
Discussion
-
-
 Discussion
Discussion